Co to jest refluks?
Wiele organicznych reakcji chemicznych trwa bardzo długo, aby zakończyć, a w celu przyspieszenia tych reakcji, ciepło jest stosowane. Związki organiczne są często lotne, mają wysoką prężność par i niską temperaturę wrzenia. Po podgrzaniu do pewnego stopnia stają się one łatwopalne i prowadzą do eksplozji. Dlatego zastosowanie ciepła musi odbywać się w specyficzny sposób, aby przezwyciężyć problem odparowania zbyt dużej ilości rozpuszczalnika i wysuszenia naczynia reakcyjnego.
Refluks polega na ogrzewaniu reakcji chemicznej przez określony czas, przy jednoczesnym ciągłym chłodzeniu wytworzonej pary z powrotem do postaci ciekłej za pomocą skraplacza. Pary powstające nad reakcją ulegają ciągłemu skraplaniu, wracając do kolby w postaci kondensatu. W ten sposób gwarantuje się, że temperatura reakcji pozostaje stała.
Reagenty w eksperymentach refluksowych mogą być stałe i ciekłe, lub obie ciecze. Temperatura, w której reakcja jest ogrzewana, zależy od temperatury wrzenia rozpuszczalników, a także od pierścienia zwrotnego (patrz poniżej).
Jeżeli substancja dodana do kolby okrągłodennej nie jest zbyt lepka, można zastosować mieszadło magnetyczne, aby zapobiec gwałtownemu uderzaniu wrzącej cieczy i zapewnić równomierne ogrzewanie. Jak pokazano na rysunku 2, gorąca płyta powinna być użyta zamiast płaszcza grzejnego, gdy używane jest mieszadło magnetyczne, ponieważ zawiera ono mieszadło magnetyczne pozwalające na automatyczne obracanie pręta podczas refluksu
Kondensator jest zawsze całkowicie wypełniony wodą, aby zapewnić skuteczne chłodzenie. Pary, które wydzielają się z ciekłej mieszaniny reakcyjnej, przechodzą z fazy gazowej z powrotem do fazy ciekłej na skutek utraty ciepła. W trakcie reakcji część rozpuszczalnika przemieszcza się w górę rurki skraplacza, a następnie skrapla się z powrotem do kolby. Powyżej tego punktu, wewnętrzny płaszcz chłodnicy wydaje się suchy. Poniżej tego punktu, rozpuszczalnik spływa z powrotem do kolby. Granica pomiędzy tymi dwoma częściami to pierścień zwrotny. Temperatura reakcji musi być tak ustawiona, aby pierścień zwrotny znajdował się tylko w jednej trzeciej do połowy wysokości chłodnicy.
Aby wiedzieć, że temperatura wrzenia została osiągnięta, wewnątrz cieczy powstają pęcherzyki pary. Jeśli szybkość ogrzewania jest zwiększona, temperatura reagentów nie zmienia się, ale szybkość, z jaką wrzącej cieczy zmienia się w postaci pary wzrasta. Wzrost ten jest spowodowany zwiększoną dostawą energii, która ułatwia więcej cząsteczek cieczy do pokonania ich oddziaływań międzycząsteczkowych, aby przejść do fazy gazowej.
Gdy mieszanina dwóch lub więcej lotnych związków jest ogrzewany, całkowita prężność pary (PT) mieszaniny równa się sumie ciśnień pary związku 1 i 2 (P1 i P2) w mieszaninie. Wielkość prężności pary wywieranej przez każdy związek zależy od prężności pary tego związku (P0) i ułamków molowych obu związków 1 i 2 obecnych w mieszaninie (X1 i X2).
Dla idealnego roztworu dwuskładnikowego prężność pary roztworu wyraża się prawem Raoulta, przedstawionym w równaniu poniżej:
PT = X1P10 + X2P20
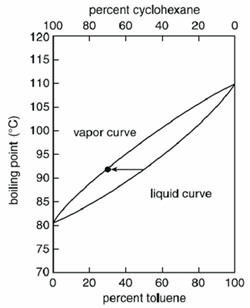
W zależności od mieszaniny temperatura wrzenia jest zmienna. Mieszaniny jednorodne wrzą w temperaturze pomiędzy punktami wrzenia czystych związków, ale dokładna wartość zależy od ilości (masy lub objętości) każdego związku.
Na przykład, mieszanina cieczy podczas wrzenia daje parę, która będzie zawierać większy procent bardziej lotnych związków. W mieszaninie cykloheksanu i toluenu, cykloheksan jest bardziej lotny między nimi i ciecz składająca się z 50 procent cykloheksanu i 50 procent toluenu będzie gotować się w temperaturze 90 ° C i daje pary składające się z 70 procent cykloheksanu i 30 procent toluenu.
Jeśli chodzi o oddzielanie związków powszechną metodą, która jest używana w chemii organicznej jest destylacja, która oddziela związki na podstawie różnic w punktach wrzenia.
W bardziej zaawansowanych eksperymentach, refluks i destylacja mogą być przeprowadzane w tym samym czasie. Na przykład, podczas refluksowania reakcji można przeprowadzić destylację w mikroskali przy użyciu specjalistycznego sprzętu. Destylacja w skali mikro ma na celu skrócenie drogi destylacji, aby zmniejszyć szansę na straty materiału w procesie.
 |
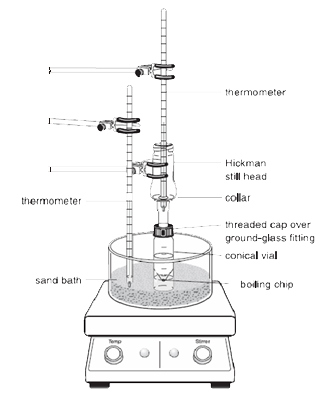 |
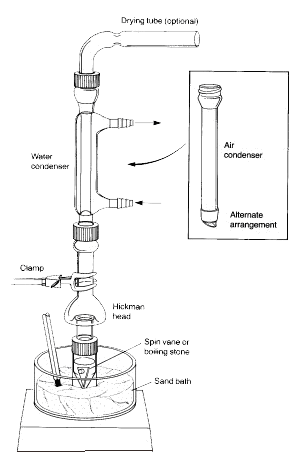
Uwaga: Pary podgrzanej cieczy unoszą się do góry i ochładzają się, by skroplić się na wewnętrznych ściankach głowicy Hickmana lub na ściankach skraplacza.
Ciecz, która spływa w dół, zbiera się w okrągłej studni na dnie still.
1.Fiolka stożkowa powinna być bezpiecznie przymocowana do głowicy destylacyjnej Hickmana i skraplacza powietrznego za pomocą nasadki kompresyjnej i metalowego zacisku. Wszystkie elementy powinny być połączone szlifem szklanym i dobrze pasować do siebie tak, aby nie występowały żadne poważne nieszczelności.
2.Wirująca łopatka powinna być umieszczona w stożkowej fiolce i skierowana w dół. Płaska przegroda i mała nasadka kompresyjna są używane do zamknięcia portu bocznego głowicy Hickmana. Cały zestaw jest umieszczony w odpowiednim otworze w Al-bloku lub łaźni piaskowej i wyśrodkowany na płycie grzejnej przed rozpoczęciem mieszania (w przeciwnym razie łopatka będzie się przewracać i obracać nieprawidłowo).
3.Głowica Hickmana i skraplacz powietrzny muszą być chłodzone mokrym ręcznikiem papierowym. Źródło ciepła powinno być tak ustawione, aby interesujący nas związek destylował powoli.
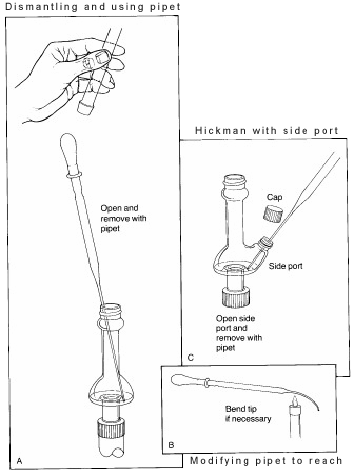
W powyższym układzie, głowica destylacyjna Hickmana działa zarówno jako skraplacz powietrza jak i naczynie zbierające kondensat dla destylacji prostej lub frakcyjnej. Głowica Hickmana może być podzielona na dwa typy: z portem i bez portu.
Przy zastosowaniu głowicy Hickmana z portem łatwiej jest zbierać frakcje. W tym celu należy otworzyć port, aby usunąć ciecz znajdującą się w studzience za pomocą pipety Pasteura (patrz 'C' na Rysunku 3).
W przypadku nieportowanej głowicy Hickmana, pipeta Pasteura jest używana do wyciągania cieczy z góry. (patrz 'A').
Jeśli używany jest skraplacz lub termometr wewnętrzny, aparat destylacyjny musi być częściowo rozmontowany w celu wykonania tych czynności. W niektórych destylatorach wewnętrzna średnica głowicy jest tak mała, że trudno jest sięgnąć pipetą pod kątem i zetknąć się z cieczą. Aby rozwiązać ten problem, końcówkę pipety należy lekko wygiąć w płomieniu.
Po usunięciu cieczy przenosimy ją do małej fiolki i zakręcamy teflonowym korkiem.Jeśli w fiolce znajduje się więcej niż jeden związek lotny, konieczne będzie rozpoczęcie destylacji od niskiej temperatury, aby najpierw oddestylować związek o niższym wrzeniu. W ten sposób można stosunkowo czysto rozdzielić związki, których różnica temperatury wrzenia wynosi co najmniej 50 oC.
Na koniec kilka słów rady:
1. Fiolka stożkowa nie powinna być wypełniona więcej niż do połowy, aby pozostawić wystarczająco dużo miejsca na zagotowanie się cieczy. W przeciwnym razie roztwór rozleje się lub przeleje, gdy zacznie wrzeć.
2. Dobre uszczelnienie pomiędzy złączami minimalizuje straty związku docelowego podczas destylacji. Zapobiega to również kapaniu związku na płytę grzejną i ewentualnemu wypadkowi przy pożarze.
3.