La fibrosi polmonare (PF), o cicatrizzazione dei polmoni, consiste in oltre 200 tipi di disturbi polmonari che sono difficili da distinguere. Tutte rientrano in una famiglia più grande di condizioni polmonari, chiamata malattia polmonare interstiziale (ILD), che comporta l’infiammazione e/o la cicatrizzazione del tessuto polmonare. Il tipo più comune di fibrosi polmonare è la fibrosi polmonare idiopatica (IPF), che si verifica quando la cicatrizzazione polmonare si verifica senza una causa nota – una condizione che colpisce uno su 200 adulti statunitensi di età superiore ai 70 anni.
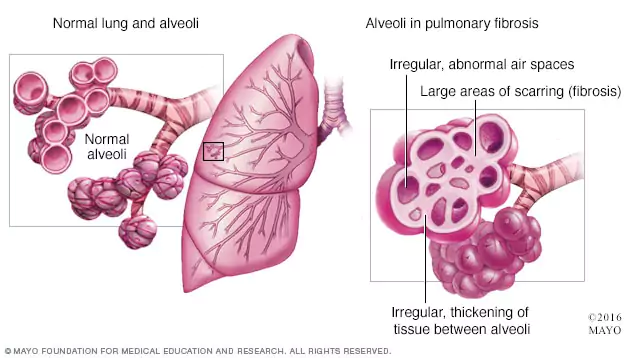
Tutti i tipi di PF comportano cicatrici e infiammazione progressive. Man mano che la cicatrizzazione aumenta, aumenta anche la mancanza di respiro, e alla fine si può sviluppare un’insufficienza polmonare. La Pulmonary Fibrosis Foundation (PFF) fa notare che non c’è modo di prevedere quanto tempo vivrà una persona con PF o IPF. L’aspettativa di vita media è di tre o cinque anni dopo la diagnosi, ma queste cifre sono ormai superate.
La progressione della malattia può essere influenzata, sia positivamente che negativamente, da una serie di fattori diversi. La Pulmonary Fibrosis Foundation (PFF) e l’University of Rochester Medical Center sottolineano diversi fattori che hanno una grande influenza sulla prognosi di un paziente con PF.
Fattori che hanno un impatto positivo sulla prognosi
- Diagnosi precoce: La diagnosi precoce gioca un ruolo importante nella prognosi di un paziente. Prima viene diagnosticata la PF o la IPF, migliori sono le possibilità di sopravvivenza. La gravità della malattia al momento della diagnosi è direttamente collegata all’aspettativa di vita.
- Trattamenti anti-carcinatura: Anche se non esiste una cura per la fibrosi polmonare, due farmaci anti-fibrotici, o trattamenti anti-carcinatura, sono stati approvati dalla Food and Drug Administration (FDA) degli Stati Uniti e hanno dimostrato clinicamente di rallentare la progressione della PF.
- Differenze individuali: La funzione polmonare può diminuire più velocemente in alcuni pazienti rispetto ad altri. Mentre molti vivono oltre i tre-cinque anni, altri sperimentano l’insufficienza respiratoria prima dei tre anni, e alcuni si ammalano molto in pochi mesi.
Le cause legate ad una prognosi negativa
Un certo numero di fattori clinici sono associati ad una prognosi peggiore, o ad un ridotto tasso di sopravvivenza, con IPF:
- Mentre l’età mediana (punto medio della fascia di età) di una diagnosi di IPF è di 66 anni, l’età più avanzata è collegata a tassi di sopravvivenza peggiori. Uno studio ha mostrato che chi ha meno di 50 anni ha una sopravvivenza mediana di 9,7 anni; chi ha 50-60 anni ha circa 5,2 anni; e chi ha 60-70 anni ha circa 2,3 anni.
- In generale, gli attuali ed ex fumatori hanno un tasso di sopravvivenza peggiore dei non fumatori con IPF.
- L’aumento della mancanza di respiro, o dispnea, ha dimostrato di essere un importante predittore di sopravvivenza dopo aver tenuto conto della gravità della malattia e di altri fattori patologici. La dispnea viene misurata cambiando i punteggi in due misure per l’IPF, come il Medical Research Council modificato (mMRC).
- I pazienti IPF con un BMI (indice di massa corporea) più basso hanno una vita più breve di quelli con BMI più alto. Lo studio di cui sopra ha mostrato una durata di vita mediana di 3,6 anni per un BMI inferiore a 25; 3,8 anni per un BMI di 25-30; e 5,8 anni per un BMI superiore a 30. Questo effetto protettivo di un basso BMI può segnalare la malnutrizione o un maggiore dispendio calorico.
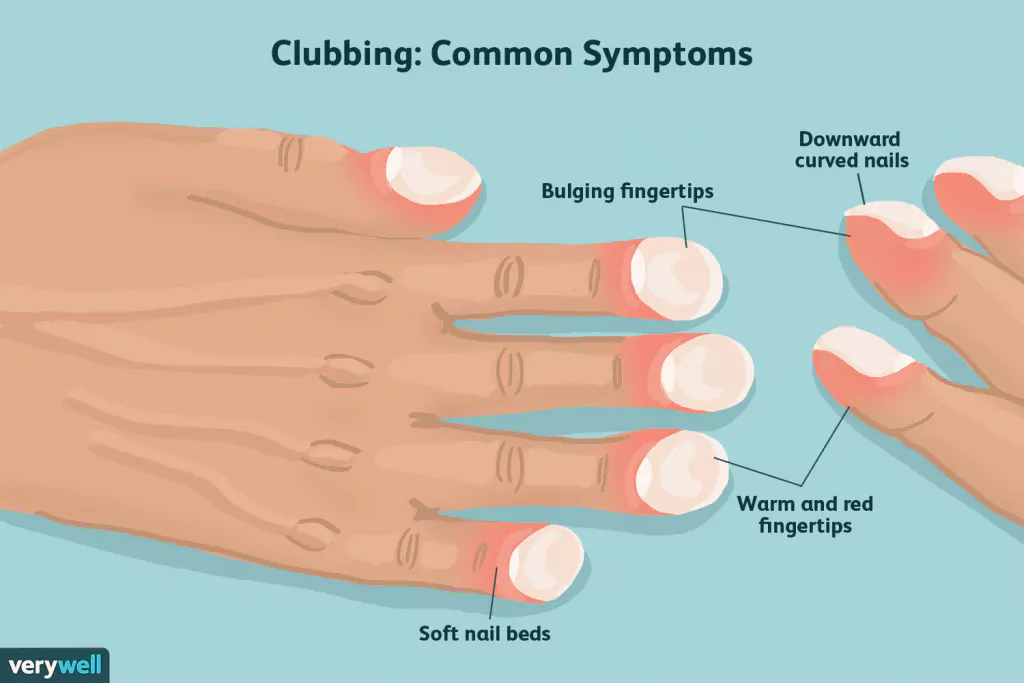
- Dopo aver tenuto conto dell’età e della storia di fumo, lo studio ha anche mostrato che le dita a bastoncello sono fortemente legate a una più breve aspettativa di vita, anche se l’American Thoracic Society (ATC) dice che sono necessari ulteriori studi.
- Il declino della funzione polmonare è legato a una prognosi peggiore e visto come una diminuzione della capacità vitale forzata (FVC), capacità polmonare totale (TLC) e capacità di diffusione dei polmoni per il monossido di carbonio (DLCO).
- Una maggiore fibrosi su una tomografia computerizzata ad alta risoluzione (HRCT) segnala la progressione dell’IPF, con un modello UIP (polmonite interstiziale abituale) generalmente legato a una peggiore aspettativa di vita.
- Malattie concomitanti, come l’ipertensione polmonare e il reflusso gastroesofageo, possono peggiorare la IPF e abbassare il tasso di sopravvivenza.
Letture aggiuntive sulla prognosi della fibrosi polmonare si possono trovare su Pulmonary Fibrosis News e sull’American Lung Association.