Che cos’è la trasposizione completa delle grandi arterie?
La trasposizione completa delle grandi arterie (TOGA) si verifica quando l’aorta, che normalmente nasce dal ventricolo sinistro e pompa sangue rosso al corpo, nasce dal ventricolo destro (1) e pompa sangue blu di ritorno dal corpo al corpo bypassando completamente i polmoni. L’arteria polmonare, che normalmente nasce dal ventricolo destro e pompa il sangue blu ai polmoni, nasce dal ventricolo sinistro (2) e manda il sangue rosso di ritorno dai polmoni direttamente ai polmoni. Essenzialmente, le grandi arterie sono invertite rispetto alle loro normali connessioni.
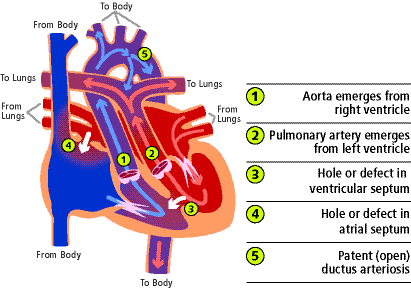
La causa del problema non è compresa. È la forma più comune di cardiopatia congenita cianotica che si presenta nel periodo neonatale. È più comune nei maschi e i bambini sono di solito di peso e dimensioni normali alla nascita. La TOGA rappresenta dal 5 al 7% di tutti i difetti cardiaci congeniti.
Ci sono diverse altre anomalie cardiache che possono verificarsi insieme alla TOGA. Il problema associato più comune è un difetto del setto ventricolare (3). Questo è un difetto o un buco nella parete che separa le due camere inferiori del cuore, i ventricoli. Ci può essere un restringimento dell’area del cuore dove il sangue fluisce verso l’arteria polmonare. Questo è chiamato ostruzione del tratto di efflusso ventricolare sinistro.
Molti di questi bambini hanno un difetto del setto atriale (4) (un foro nella parete che separa le due camere superiori del cuore) e/o un dotto arterioso pervio (5). Questo è un normale canale di nascita tra l’aorta e l’arteria polmonare presente alla nascita che può non chiudersi in presenza di altri problemi cardiaci.
Quali sono gli effetti di questo difetto sulla salute di mio figlio?
Quando un bambino ha la TOGA, ci sono due circuiti separati di flusso di sangue invece di uno collegato. Il sangue blu che ritorna dal corpo è pompato indietro verso il corpo e il sangue rosso che ritorna dai polmoni è pompato indietro verso i polmoni. Come risultato, il bambino sviluppa un colore blu, chiamato cianosi, poco dopo la nascita. Il colore blu può essere notato meglio nelle labbra o sotto le unghie. In un bambino con cianosi legata al cuore, il colore blu non migliora con l’uso di ossigeno.
Se questa situazione dovesse continuare, il bambino potrebbe presto morire per mancanza di ossigeno nel corpo. L’unico modo in cui un bambino con TOGA può sopravvivere dopo la nascita è se c’è un modo per il sangue rosso e blu di mescolarsi insieme all’interno del cuore in modo che un po’ di sangue rosso venga pompato fuori al corpo. Un difetto del setto atriale e/o un dotto arterioso pervio di solito permettono abbastanza ossigeno per permettere al bambino di sopravvivere fino a quando un intervento più definitivo può essere eseguito.
Alcuni bambini con TGA hanno anche un foro tra le camere inferiori del cuore chiamato difetto del setto ventricolare. Se questo è presente, può verificarsi una mescolanza di sangue tale che il bambino può non apparire affatto cianotico e può effettivamente ammalarsi con sintomi di insufficienza cardiaca congestizia a causa del flusso di sangue extra che va ai polmoni. Allora il bambino avrà sintomi di scarsa alimentazione, scarso aumento di peso, sudorazione e respirazione veloce o affannosa.
Infine, ci può essere un restringimento dell’area che porta il ventricolo sinistro all’arteria polmonare chiamato ostruzione del tratto di efflusso del ventricolo sinistro. In questa situazione, anche se c’è il foro per il sangue da mescolare, la quantità totale di flusso di sangue che va nei polmoni è ridotta. Il grado di restringimento varia e può essere lieve all’inizio, ma può peggiorare col tempo. Man mano che il restringimento aumenta, la colorazione del bambino diventerà più cianotica (blu).
La gravità dei sintomi dipende dalla quantità di sangue rosso e blu che si mischia e dalla presenza o assenza di ostruzione al flusso di sangue dal ventricolo sinistro. Il tipo e i tempi dell’operazione dipendono dalla combinazione di difetti che accompagnano il problema primario della TGA.
I bambini con TGA possono sviluppare una malattia vascolare polmonare precoce. Si tratta di un aumento della pressione nei vasi sanguigni polmonari che causa cambiamenti che rendono difficile per loro accettare un flusso di sangue a bassa pressione. Questi cambiamenti possono verificarsi già a poche settimane di vita e tendono a verificarsi più frequentemente nei bambini che hanno difetti del setto ventricolare oltre alla TGA. La chirurgia correttiva precoce minimizza le possibilità di sviluppo di un’elevata resistenza vascolare polmonare in questi bambini.
Come viene diagnosticato questo difetto?
Caratteristiche cliniche: Come descritto sopra, i bambini con TOGA completa hanno livelli di ossigeno nel sangue più bassi dal momento della nascita. Il colore blu si vede nelle labbra e sotto i letti delle unghie e può essere abbastanza difficile da rilevare solo guardando il bambino. Si sviluppano segni di insufficienza congestizia, compresi i sintomi dell’insufficienza cardiaca congestizia, tra cui l’eccessiva sudorazione (un sudore freddo e umido spesso notato durante l’alimentazione); scarsa alimentazione, lento aumento di peso, irritabilità o letargia, e/o respirazione rapida di solito si sviluppano durante il periodo neonatale.
Reperti fisici: La TOGA completa viene solitamente diagnosticata a 24-48 ore dalla nascita per la presenza di cianosi che è moderata o grave nella maggior parte dei casi. Il secondo suono cardiaco è forte e singolo. Ci può essere o meno un soffio a seconda della presenza di un difetto del setto ventricolare. Se il bambino è in insufficienza cardiaca congestizia, la frequenza respiratoria sarà veloce e il fegato sarà ingrossato.
Test medici: I livelli di ossigeno nel sangue possono essere misurati con un test di saturazione dell’ossigeno o con un esame del sangue. A volte il bambino viene messo in una tenda ad ossigeno e gli viene dato ossigeno al 100% per vedere se i livelli di ossigeno nel sangue aumentano. Se i livelli di ossigeno aumentano significativamente, ciò suggerisce che il basso livello di ossigeno è dovuto a un problema polmonare invece che a un problema cardiaco.
Di solito si fa anche un elettrocardiogramma e una radiografia del petto. Il difetto è diagnosticato da un test cardiaco chiamato ecocardiogramma o ecografia cardiaca. L’ecocardiogramma usa le onde sonore per formare un’immagine delle valvole e delle camere all’interno del cuore. È sicuro e indolore e i risultati del test sono disponibili immediatamente. In alcuni casi, la diagnosi viene fatta prima della nascita durante un’ecografia fetale. La prima volta che il test può essere usato per diagnosticare questo problema è quando la madre è alla 18° settimana di gravidanza.
Un altro test del cuore chiamato cateterismo cardiaco sarà necessario se l’ecocardiogramma non è completamente chiaro sul problema cardiaco o se sono presenti ulteriori anomalie che possono influenzare il modo in cui il chirurgo sistemerebbe il difetto. Durante questa procedura, i cateteri (sottili tubi di plastica) vengono inseriti nei grandi vasi sanguigni situati nella zona inguinale e guidati delicatamente nel cuore. Il contrasto o “colorante” viene quindi inserito nel cuore in modo da poter scattare immagini a raggi X. Si ottengono anche misure di pressione e livelli di ossigeno. È più complesso dell’ecocardiogramma, ma è considerato molto sicuro. I bambini sono sedati e gli vengono dati degli antidolorifici durante il test. I risultati sono più spesso disponibili il giorno stesso.
Cliniche
La cura e i servizi per i pazienti con questo problema sono forniti nelle cliniche del Cuore Congenito e della Chirurgia Cardiovascolare dell’Università del Michigan Health System a Ann Arbor.
Come viene trattato il problema?
Trattamenti Parte 1 | Trattamenti Parte 2
Lupinetti FM, Bove EL, Snider AR, Callow LB, Meliones JN, Crowley DC, Beekman RH, Minich LL, Rosenthal A: Sopravvivenza a medio termine e risultati funzionali dopo riparazione arteriosa per trasposizione delle grandi arterie. J Thorac Cardiovasc Surg 1992;103:421-7.
Bove EL, Beekman RH, Snider AR, Rocchini AP, Dick M, Crowley DC, Serwer GS, Rosenthal A: riparazione arteriosa per trasposizione e grande difetto del setto ventricolare nella prima infanzia. Circulation l988;78 (Suppl III):III-26-III-3l.
Scritto da: Louise Callow RN, MSN (12/99)
Rivista settembre 2012